GPMelt for Thermal Proteome Profiling (TPP)
1 I. Introduction
GPMelt was originally developed to analyse Temperature - Range Thermal Proteome Profiling (TPP-TR) datasets.
While GPMelt’s application is not restricted to TPP datasets, this tutorial will be based on TPP.
We begin by introducing you to TPP, its data and challenges it addresses.
2 II. Protein thermal stability
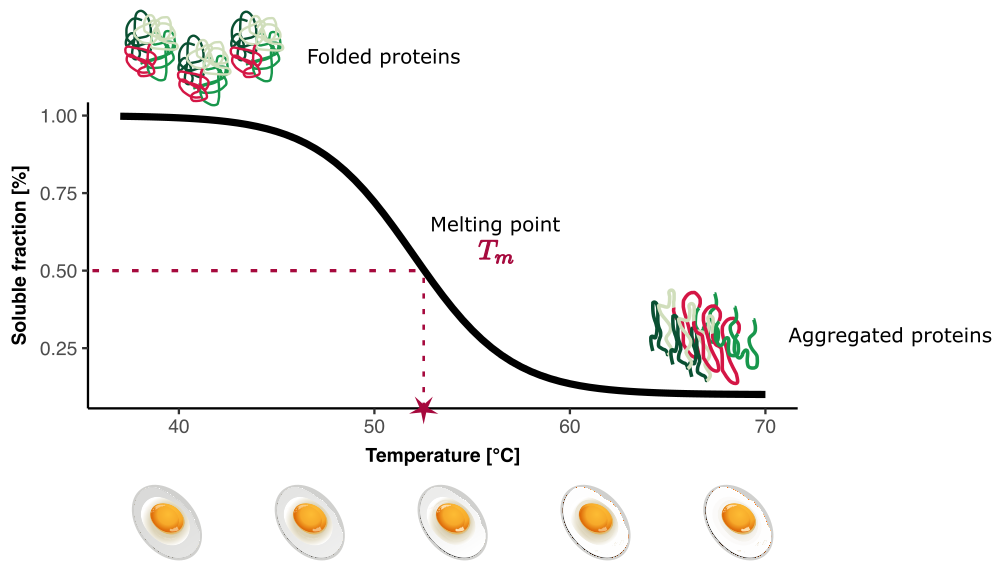
With TPP, we aim to study the thermal stability of proteins. Even if you’re not familiar with this concept yet, you’ve likely experienced it. For example, when cooking an egg, you may have noticed that the egg white starts off transparent and fluid, but then turns white and firm. What’s happening here? The proteins in the egg white are being denatured by heat—this is what we call the heat-induced denaturation process.
In TPP, we measure how proteins in a bilogical sample behave when heated. Figure 1 illustrates the theoretical melting behaviour of a protein in a tube. At physiological temperatures, proteins in the sample are mainly in a folded configuration. As the temperature increases, the atomic bonds that maintain this structure are disrupted, and the proteins begin to unfold. As they unfold, new bonds can form between the unfolded proteins, leading to protein aggregation.
In TPP, we measure the fraction, relative to the lowest measured temperature, of proteins in the folded form. We call this the soluble fraction.
An important quantity to characterise the protein thermal stability is the melting point \(T_m\), which is the temperature at which half of the proteins in the sample have denatured.
3 III. Changes in thermal stability
When studying protein thermal stability, the most informative approach is to compare the melting behaviour of a given protein under different conditions. This is particularly useful for detecting drug targets and off-targets. The concept is simple: if a compound (e.g. a drug) interacts with a protein, the interaction will affect the biophysical properties of the protein. This, in turns, lead to a thermal stabilisation or destabilisation of the protein. This means that the protein becomes more or less resistant to thermal stress. In drug development, the goal is to design small compounds that are as specific as possible to their targets, as any unwanted interactions could lead to side effects.
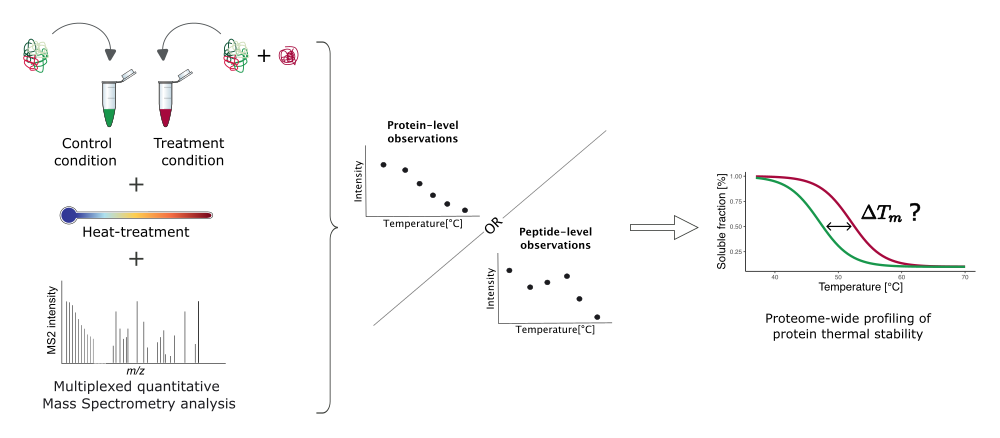
To investigate drug binding on a proteome-wide scale, (Savitski et al. 2014) proposed to combine the Cellular Thermal Shift Assay (CETSA) (Molina et al. 2013) with mulitplexed quantitative Mass Spectrometry (MS) analysis. This method, known as TPP, theoretically enables the detection of all proteins whose thermal stability changes upon treatment.
Figure 2 summarises the principles of TPP experiments.
4 IV. TPP dataset analysis based on the melting point definition
In Figure 1, we described the theoretical melting behaviour of a protein, which is characterised by its sigmoidal shape and melting point \(T_m\). When a drug stabilises the protein (as depicted in Figure 2), we expect the melting point of the protein in the treatment condition (red curve) to be higher than in the control condition (green curve). Therefore, comparing the estimated melting points between control and treatment conditions should be sufficient to determine whether the protein has been stabilised or destabilised by the treatment.
This is the principle on which previous analyses, such as the \(T_m\) analysis (Savitski et al. 2014) and the NPARC method (Childs et al. 2019), were based.
5 V. The presence of non-sigmoidality
These previous analyses worked well for sigmoidal melting curves, similar to the ones illustrated in Figure 1.
But why did we use a sigmoid shape before? The sigmoidal shape can be explained by thermodynamic models describing purified proteins in a test tube.
However, TPP samples are much more complex: they can consist of intact cells, cell lysates, tissues, or biological fluids. As a result, many additional cellular mechanisms must be considered when describing protein melting behaviours.
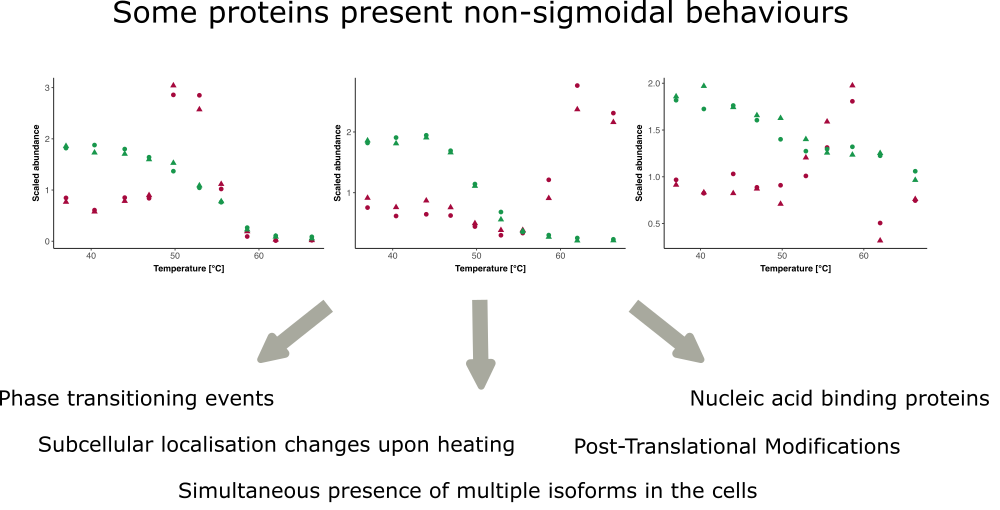
While researchers do not yet fully understand all these mechanisms, they have observed that a non-negligeable fraction of proteins in their TPP datasets do not exhibit sigmoidal melting behaviour. They have also begun to propose biological hypotheses for these behaviours (see Figure 3).
6 VI. Incorporating non-conventional melting curves in the analysis.
Pioneering the use of Gaussian Processes, (Fang et al. 2021) introduced a new model, called the Bayesian semi-parametric model, for TPP dataset analysis. The use of Gaussian processes makes it possible to incorporate any melting curve shapes in the analysis.
Building GPMelt on the same principle of non-conventional melting curves inclusion, we propose a new method which is more robust to outliers than the Bayesian semi-parametric model.
Our method is also more versatile, dealing with multiple treatment conditions at once, and applicable to more complex TPP protocols.
Follow the tutorial to learn how to use GPMelt and discover the additional cool features it offers!
Also, keep in mind that TPP datasets are not the only type of data that can be analysed with GPMelt. This method works for any time-series data where you wish to compare shapes between conditions, especially when dealing with multiple conditions and/or replicates!